介紹 phylogram 這個 R 語言的演化樹資料格式整合套件的使用方式以及實務範例。
系統發生樹(phylogenetic tree)又稱為演化樹或進化樹(evolutionary tree),是演化生物學家常需要繪製的圖形,而 R 中有許多的套件都可以用來畫這類的圖,例如 ape、phangorn 與 phytools,但是這些工具所使用的資料格式並沒有整合在一起。
比較常見的資料格式有 phylo 與 dendrogram 這兩種。phylo 是以矩陣方式來儲存資料的格式,效率較高、功能較陽春,採用這種格式的套件有 ape、phangorn 與 Phytools 等;dendrogram 則是以巢狀列表(nested list)的方式儲存資料,雖然效率較低,但是功能較豐富、直覺好操作,同時亦可使用一般列表的操作函數來處理,所以有很多工具(例如 dendextend)都使用這種格式。
phylogram 是一個整合 phylo 與 dendrogram 兩種格式的工具套件,可以用來處理演化樹的格式轉換、資料管理、匯入與匯出等動作,同時也支援 Newick 格式,以下是 phylogram 套件的使用教學。
phylogramphylogram 套件可以從 CRAN 官方套件庫下載與安裝:
# 安裝 phylogram 套件 install.packages("phylogram")
若想要安裝最新的開發中版本,則可先安裝 devtools、kmer 與 dendextend 這幾個必要的套件之後,再從 GitHub 上面下載並安裝 phylogram:
# 從 GitHub 下載並安裝 phylogram 套件 devtools::install_github("ropensci/phylogram", build_vignettes = TRUE)
載入 phylogram 套件:
# 載入 phylogram 套件 library(phylogram)
Newick 是許多樹狀圖編輯軟體都支援的一種樹狀圖格式,而 phylogram 套件所提供的 read.dendrogram 函數就可以用來匯入 Newick 格式的樹狀圖:
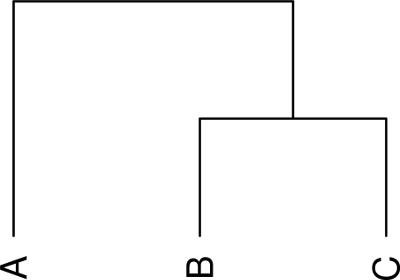
# 匯入 Newick 格式樹狀圖 my.tree.1 <- read.dendrogram(text = "(A,(B,C));")
dendrogram 的樹狀圖可以直接使用 plot 將它畫出來:
# 繪製樹狀圖 plot(my.tree.1, yaxt = "n")
繪製出來的圖形會像這樣:
若想要將 dendrogram 格式的樹狀圖匯出成為 Newick 格式的文字,可以使用 phylogram 套件所提供的 write.dendrogram 函數,以下是一個簡單的範例。
# 將樹狀圖匯出為 Newick 格式 write.dendrogram(my.tree.1, edges = FALSE)
[1] "(A,(B,C));"
若要直接從 Newick 格式的檔案中讀取樹狀圖,作法也都大同小異,只要使用 file 參數指定來源檔案名稱即可:
# 從檔案匯入 Newick 格式樹狀圖 my.tree.2 <- read.dendrogram(file = "my_tree.dnd")
若要將樹狀圖匯出至檔案,則在呼叫 write.dendrogram 時以 file 參數指定輸出的檔案名稱:
# 將樹狀圖匯出為 Newick 格式的檔案 write.dendrogram(my.tree.2, file = "output.dnd", edges = FALSE)
phylogram 套件所提供的 as.phylo.dendrogram 與 as.dendrogram.phylo 函數可以用來處理 phylo 與 dendrogram 格式之間的轉換,但是除了 phylogram 套件之外,其他的套件也有可能會提供相同名稱的函數,若要確保能夠呼叫到 phylogram 套件所提供的轉換函數,在使用時可以加上命名空間的標示:
# 從 dendrogram 轉換為 phylo my.phylo.1 <- phylogram::as.phylo.dendrogram(my.tree.1) # 從 phylo 轉換為 dendrogram my.dend.1 <- phylogram::as.dendrogram.phylo(my.phylo.1)
phylogram 套件提供了一些可以用來編輯 dendrogram 的函數,prune 可以用來移除符合條件的節點,ladder 函數則可以用來調整節點的順序,方便畫圖。
若以 R 基本的巢狀列表(nested list)建立演化樹的時候,可以使用 remidpoint 函數自動修正各個節點的 midpoint、members、leaf 等屬性,而節點的高度則可使用 reposition 或 as.cladogram 來設定。
# 建立基本的巢狀列表 my.tree.3 <- list(1, list(2, 3)) # 設定各個節點的 midpoint、members、leaf 等屬性 my.tree.3 <- remidpoint(my.tree.3) # 設定各個節點的高度屬性 my.tree.3 <- as.cladogram(my.tree.3)
在進行 dendrogram 的各種編輯動作時,可以運用 dendrapply 或 lapply 函數,將自訂的處理函數一次套用至所有的節點上。
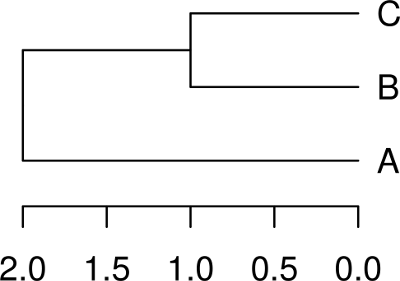
# 建立自訂處理函數 set_label <- function(node) { if (is.leaf(node)) attr(node, "label") <- LETTERS[node] return (node) } # 遞迴處理每個節點 my.tree.3 <- dendrapply(my.tree.3, set_label) # 繪製樹狀圖 plot(my.tree.3, horiz = TRUE)
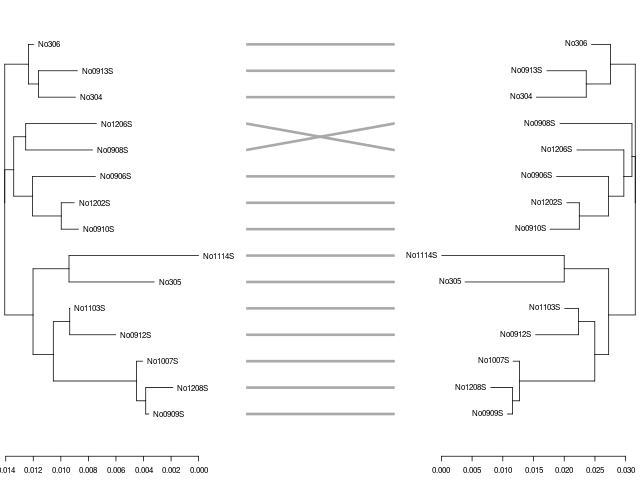
若要產生兩棵演化樹的比較圖,可以使用 dendextend 套件的 tanglegram 繪圖函數來處理,但這個繪圖函數只接受 dendrogram 格式,若遇到 phylo 的時候就要使用 phylogram 套件來轉換。
以下示範使用 k-mer 與 Kimura 1980 兩種不同距離計算方式,產生兩棵演化樹,比較它們的差異。
首先安裝 dendextend 這個繪圖用的套件,以及 kmer 這個計算 k-mer 距離的套件:
# 安裝 dendextend 與 kmer 套件 install.packages(c("dendextend", "kmer"))
其實這裡還需要一個 ape 演化樹分析套件,不過這個套件在安裝 phylogram 套件的時候,就已經自動安裝好了,所以不用手動安裝。
首先載入必要的套件以及 woodmouse 這個示範用的資料集。
# 載入套件與資料 library(ape) library(kmer) data(woodmouse)
使用 k-mer 與 Kimura 1980 演算法產生兩個距離矩陣:
# 計算距離矩陣 woodmouse.dist <- ape::dist.dna(woodmouse, model = "K80") woodmouse.kdist <- kdistance(woodmouse, k = 6)
以近鄰結合法(neighbor-joining method)建立演化樹:
# 近鄰結合法建立演化樹 phy1 <- ape::nj(woodmouse.dist) phy2 <- ape::nj(woodmouse.kdist)
重新排列節點順序,可讓畫出的演化樹更好閱讀:
# 重新排列節點順序 phy1 <- ape::ladderize(phy1) phy2 <- ape::ladderize(phy2)
將 phylo 的樹轉換為 dendrogram 的樹:
# 將 phylo 物件轉換為 dendrogram 物件 dnd1 <- phylogram::as.dendrogram(phy1) dnd2 <- phylogram::as.dendrogram(phy2)
呼叫 tanglegram 繪製樹狀圖:
# 繪製樹狀圖 dndlist <- dendextend::dendlist(dnd1, dnd2) dendextend::tanglegram(dndlist, fast = TRUE, margin_inner = 5)
參考資料:rOpenSci、CRAN、GitHub、kmer vignettes、dendextend vignettes、CRAN Task View
{kind=link}
{kind=link}
{kind=link}