介紹如何在 R 中匯入 phyloseq 微生物分析套件用的資料。
phyloseq 資料匯入工具phyloseq 提供了許多的資料匯入與轉換工具,可將 R 中各種形式的資料轉換為 phyloseq 格式資料。
以下是一些常用的資料轉換工具:
otu_tablematrix 格式的 OTU 表格轉換為 otu_table 格式。sample_datadata.frame 格式的後設資料(metadata)轉換為 sample_data 格式,若要與 OTU 表合併至同一個 phyloseq 物件,則要注意列名稱必須與 OTU 表格的樣本名稱吻合。tax_tablematrix 格式的分類(taxonomy)表格轉換為 taxonomyTable 格式,若要與 OTU 表合併至同一個 phyloseq 物件,則要注意列名稱必須與 OTU 表格的樣本名稱吻合。phyloseqotu_table 加上 sample_data、taxonomyTable、phylo 或 XStringSet 物件合併成一個 phyloseq 物件。merge_phyloseqphyloseq 物件合併成單一個 phyloseq 物件。關於這些基本函數的操作方法,可參閱以下幾個函數的文件:
# 載入 phyloseq 套件 library(phyloseq) # 匯入資料相關主要文件 ?phyloseq ?otu_table ?sample_data ?tax_table
phyloseq 物件為了示範將最基本的 R 原始資料轉換為 phyloseq 格式的資料,我們先產生一個數值 matrix 的 OTU 表格:
# 產生 OTU 表格 set.seed(1) otu.matrix <- matrix(sample(1:100, 100, replace = TRUE), nrow = 10, ncol = 10) # 設定行列名稱 rownames(otu.matrix) <- paste0("OTU", 1:nrow(otu.matrix)) colnames(otu.matrix) <- paste0("Sample", 1:ncol(otu.matrix)) # 查看 OTU 表格 otu.matrix
Sample1 Sample2 Sample3 Sample4 Sample5 Sample6 Sample7 Sample8 Sample9 Sample10 OTU1 68 97 89 74 44 2 81 13 73 93 OTU2 39 85 37 42 25 45 100 22 87 34 OTU3 1 21 34 38 70 18 13 93 83 10 OTU4 34 54 89 20 39 22 40 28 90 1 OTU5 87 74 44 28 51 78 89 48 48 43 OTU6 43 7 79 20 42 65 48 33 64 59 OTU7 14 73 33 44 6 70 89 45 94 26 OTU8 82 79 84 87 24 87 23 21 96 15 OTU9 59 85 35 70 32 70 84 31 60 58 OTU10 51 37 70 40 14 75 29 17 51 29
接著產生分類表格:
# 產生分類表格 tax.matrix <- matrix(sample(letters, 70, replace = TRUE), nrow = nrow(otu.matrix), ncol = 7) # 設定行列名稱 rownames(tax.matrix) <- rownames(otu.matrix) colnames(tax.matrix) <- c("Domain", "Phylum", "Class", "Order", "Family", "Genus", "Species") # 查看分類表格 tax.matrix
Domain Phylum Class Order Family Genus Species OTU1 "x" "a" "i" "s" "o" "d" "b" OTU2 "j" "s" "e" "l" "g" "b" "b" OTU3 "p" "z" "n" "q" "o" "p" "s" OTU4 "u" "c" "q" "i" "t" "a" "q" OTU5 "l" "k" "c" "r" "y" "l" "b" OTU6 "g" "a" "v" "x" "x" "g" "k" OTU7 "x" "n" "c" "v" "s" "i" "c" OTU8 "u" "v" "g" "q" "u" "z" "c" OTU9 "v" "f" "n" "r" "p" "m" "n" OTU10 "h" "t" "c" "w" "s" "z" "s"
這我們所產生的 otu.matrix 與 tax.matrix 只是普通的 R 數值矩陣而已,接下來我們要將這些資料轉換為 phyloseq 格式的資料:
# 將 OTU 表格轉為 otu_table 格式 otu.table <- otu_table(otu.matrix, taxa_are_rows = TRUE) # 將分類表格轉為 taxonomyTable 格式 tax.table <- tax_table(tax.matrix) # 建立 phyloseq 物件 physeq <- phyloseq(otu.table, tax.table) physeq
phyloseq-class experiment-level object otu_table() OTU Table: [ 10 taxa and 10 samples ] tax_table() Taxonomy Table: [ 10 taxa by 7 taxonomic ranks ]
這樣就成功將一般的 R 資料轉換為 phyloseq 格式的物件了。
這裡我們只使用最基本的 OTU 表格與分類表格來建立 phyloseq 物件,實務上操作時如果還有其他的資料(例如 sample_data 格式的後設資料),也可以一併放進去,合併成單一個 phyloseq 格式,可讓分析流程更簡便。
將資料都整理成單一個 phyloseq 物件之後,就可以使用 phyloseq 套件的工具或其他延伸套件工具,進行各種微生物相的分析了,例如畫出豐富度的圖形:
# 豐富度圖形 library(ggplot2) plot_bar(physeq, fill = "Family") + scale_fill_brewer(palette = "Set1")
通常在分析微生物的資料時,都會有一些後設資料以及演化樹,這裡我們還是以隨機的方式,產生一些測試用的資料作為示範。
# 產生後設資料 sample.data.df <- data.frame( Location = sample(LETTERS[1:4], size=nsamples(physeq), replace=TRUE), Depth = sample(50:1000, size=nsamples(physeq), replace=TRUE), row.names = sample_names(physeq), stringsAsFactors = FALSE ) # 將後設資料轉為 sample_data 格式 sample.data <- sample_data(sample.data.df) sample.data
Sample Data: [10 samples by 2 sample variables]:
Location Depth
Sample1 A 768
Sample2 C 549
Sample3 D 810
Sample4 D 721
Sample5 A 278
Sample6 B 472
Sample7 D 470
Sample8 B 189
Sample9 D 175
Sample10 B 575
接著使用 ape 套件的 rtree 函數,隨機產生一個測試用的演化樹資料:
# 隨機產生演化樹 library("ape") phy.tree <- rtree(ntaxa(physeq), rooted = TRUE, tip.label = taxa_names(physeq)) plot(phy.tree)
準備好後設資料與演化樹之後,可以使用 merge_phyloseq 函數將這些資料併入計有的 phyloseq 物件中:
# 加入後設資料與演化樹 physeq.1 <- merge_phyloseq(physeq, sample.data, phy.tree) physeq.1
phyloseq-class experiment-level object otu_table() OTU Table: [ 10 taxa and 10 samples ] sample_data() Sample Data: [ 10 samples by 2 sample variables ] tax_table() Taxonomy Table: [ 10 taxa by 7 taxonomic ranks ] phy_tree() Phylogenetic Tree: [ 10 tips and 9 internal nodes ]
或是重新建立一個新的 phyloseq 物件也可以:
# 重新建立 phyloseq 物件 physeq.2 <- phyloseq(otu.table, tax.table, sample.data, phy.tree) physeq.2
phyloseq-class experiment-level object otu_table() OTU Table: [ 10 taxa and 10 samples ] sample_data() Sample Data: [ 10 samples by 2 sample variables ] tax_table() Taxonomy Table: [ 10 taxa by 7 taxonomic ranks ] phy_tree() Phylogenetic Tree: [ 10 tips and 9 internal nodes ]
不管是併入既有的 phyloseq 物件或是重新建立一個新的,產生的物件都是相同的:
# 檢查兩個 phyloseq 物件是否相同 identical(physeq.1, physeq.2)
[1] TRUE
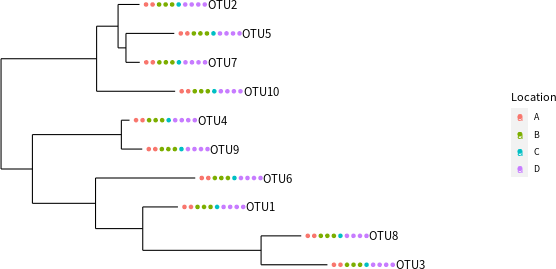
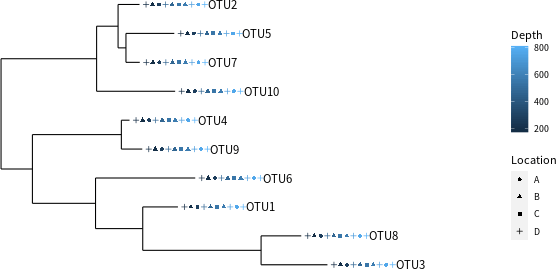
將所有的資料合併成一個 phyloseq 物件之後,就可以利用 phyloseq 套件所提供的工具繪製一些圖形,例如搭配後設資料繪製演化樹:
# 搭配後設資料繪製演化樹 plot_tree(physeq.1, color = "Location", label.tips = "taxa_names")
# 搭配後設資料繪製演化樹 plot_tree(physeq.1, color = "Depth", shape = "Location", label.tips = "taxa_names")
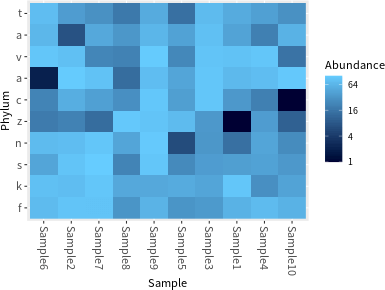
若要繪製熱圖(heatmap),可以使用 plot_heatmap 函數:
# 繪製熱圖(heatmap) plot_heatmap(physeq.1, taxa.label = "Phylum")
BIOM 是一種儲存生物樣本的檔案格式,若要從 BIOM 格式匯入資料,可以使用 import_biom 函數,以下是一個簡單的範例:
# 取得 phyloseq 套件附帶的範例檔案路徑 rich_dense_biom <- system.file("extdata", "rich_dense_otu_table.biom", package="phyloseq") rich_sparse_biom <- system.file("extdata", "rich_sparse_otu_table.biom", package="phyloseq") min_dense_biom <- system.file("extdata", "min_dense_otu_table.biom", package="phyloseq") min_sparse_biom <- system.file("extdata", "min_sparse_otu_table.biom", package="phyloseq") treefilename <- system.file("extdata", "biom-tree.phy", package="phyloseq") refseqfilename <- system.file("extdata", "biom-refseq.fasta", package="phyloseq") # 從 BIOM 檔案匯入資料 physeq.3 <- import_biom(rich_dense_biom, treefilename, refseqfilename, parseFunction = parse_taxonomy_greengenes)
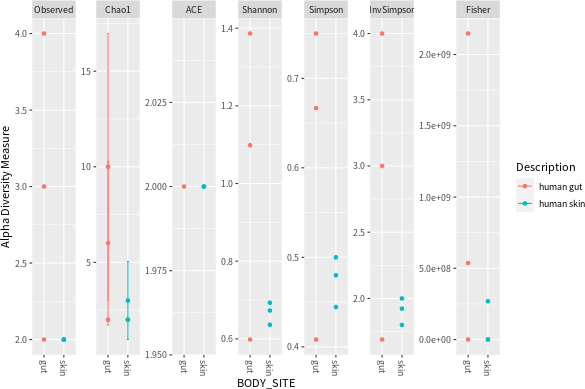
import_biom 會直接將匯入的資料轉換為 phyloseq 物件,接著就可以按照一般的方式使用資料:
# 繪製豐富度圖形 plot_richness(physeq.3, x = "BODY_SITE", color = "Description")
phyloseq 所提供的 import_qiime 函數可以用來匯入原生的 QIIME 檔案:
# 取得 phyloseq 套件附帶的範例檔案路徑 otufile <- system.file("extdata", "GP_otu_table_rand_short.txt.gz", package="phyloseq") mapfile <- system.file("extdata", "master_map.txt", package="phyloseq") trefile <- system.file("extdata", "GP_tree_rand_short.newick.gz", package="phyloseq") rsfile <- system.file("extdata", "qiime500-refseq.fasta", package="phyloseq") # 從 QIIME 檔案匯入資料 physeq.4 <- import_qiime(otufile, mapfile, trefile, rsfile)
由於 QIIME 目前也支援匯出 BIOM 檔案,所以在實務上可以自己選擇比較方便的方式匯入資料。
若要從各種其他類型的檔案格式(例如 mothur)匯入資料,可以參考 phyloseq 的 import 函數文件:
# 查看 phyloseq 匯入資料相關文件
?import
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}