整理 R 語言中幾個重要的譜系學套件,並簡單介紹其基本使用方式。
在眾多的譜系學套件中,最重要的核心就是 ape 這個套件,如果要安裝這個套件,可直接從 R 官方的 CRAN 套件庫下載安裝:
# 安裝 ape 套件 install.packages("ape")
在 R 官方的 CRAN Task View 網頁中,有整理出非常多譜系學(Phylogenetics)相關的 R 套件,如果希望一次將所有譜系學相關的套件都安裝起來,可以使用 ctv(CRAN Task View)這個套件來自動安裝:
# 安裝並載入 ctv 套件 install.packages('ctv') library('ctv') # 安裝 Phylogenetics 這個 CRAN Task View 下的所有套件 install.views('Phylogenetics') # 更新 Phylogenetics 這個 CRAN Task View 下的所有套件 update.views('Phylogenetics')
ape 套件ape 這個套件的功能非常豐富,使用前先載入套件:
# 載入 ape 套件 library(ape)
接下來就可以開始使用 ape 套件的各項功能了。
ape 所提供的 read.dna 函數可以從檔案讀取 DNA 的序列資料,常見的幾種文字格式它都支援,例如讀取普通的 sequential 格式的 DNA 序列資料:
# 建立 sequential 格式的 DNA 序列資料 cat("3 40", "No305 NTTCGAAAAACACACCCACTACTAAAANTTATCAGTCACT", "No304 ATTCGAAAAACACACCCACTACTAAAAATTATCAACCACT", "No306 ATTCGAAAAACACACCCACTACTAAAAATTATCAATCACT", file = "dna1.txt", sep = "n") # 讀取 sequential 格式的 DNA 序列資料 dna1 <- read.dna("dna1.txt", format = "sequential")
讀取 interleaved 格式的 DNA 序列資料:
# 建立 interleaved 格式的 DNA 序列資料 cat("3 40", "No305 NTTCGAAAAA CACACCCACT", "No304 ATTCGAAAAA CACACCCACT", "No306 ATTCGAAAAA CACACCCACT", " ACTAAAANTT ATCAGTCACT", " ACTAAAAATT ATCAACCACT", " ACTAAAAATT ATCAATCACT", file = "dna2.txt", sep = "n") # 讀取 interleaved 格式的 DNA 序列資料 dna2 <- read.dna("dna2.txt")
讀取 clustal 格式的 DNA 序列資料:
# 建立 clustal 格式的 DNA 序列資料 cat("CLUSTAL (ape) multiple sequence alignment", "", "No305 NTTCGAAAAACACACCCACTACTAAAANTTATCAGTCACT", "No304 ATTCGAAAAACACACCCACTACTAAAAATTATCAACCACT", "No306 ATTCGAAAAACACACCCACTACTAAAAATTATCAATCACT", " ************************** ****** ****", file = "dna3.txt", sep = "n") # 讀取 clustal 格式的 DNA 序列資料 dna3 <- read.dna("dna3.txt", format = "clustal")
讀取 FASTA 格式的 DNA 序列資料:
# 建立 FASTA 格式的 DNA 序列資料 cat(">No305", "NTTCGAAAAACACACCCACTACTAAAANTTATCAGTCACT", ">No304", "ATTCGAAAAACACACCCACTACTAAAAATTATCAACCACT", ">No306", "ATTCGAAAAACACACCCACTACTAAAAATTATCAATCACT", file = "dna4.fas", sep = "n") # 讀取 FASTA 格式的 DNA 序列資料 dna4 <- read.dna("dna4.fas", format = "fasta")
資料格式並不會影響資料的內容,不管用那一種資料格式,讀取進來之後的資料都是相同的,我們可以使用 identical 來驗證:
# 檢查各種資料格式讀取的結果 identical(dna1, dna2) identical(dna1, dna3) identical(dna1, dna4)
若要從 ZIP 壓縮檔案中直接讀取 DNA 序列的資料,只要先用 unz 函數解壓縮之後,再交給 read.dna 讀取即可:
# 建立 ZIP 壓縮檔 zip("dna5.fas.zip", "dna4.fas") # 從 ZIP 壓縮檔中讀取 DNA 序列資料 dna5 <- read.dna(unz("dna5.fas.zip", "dna4.fas"), "fasta")
若要讀取 GZIP 壓縮格式的 DNA 序列,則可先用 gzfile 解壓縮,然後再以 read.dna 讀取:
# 建立 GZ 壓縮檔 gz.con <- gzfile("dna6.fas.gz", "wt") cat(">No305", "NTTCGAAAAACACACCCACTACTAAAANTTATCAGTCACT", ">No304", "ATTCGAAAAACACACCCACTACTAAAAATTATCAACCACT", ">No306", "ATTCGAAAAACACACCCACTACTAAAAATTATCAATCACT", file = gz.con, sep = "n") close(gz.con) # 從 GZ 壓縮檔中讀取 DNA 序列資料 dna6 <- read.dna(gzfile("dna6.fas.gz"), "fasta")
壓縮格式也同樣不會對資料本身產生影響:
# 檢查各種資料格式讀取的結果 identical(dna5, dna4) identical(dna6, dna4)
在建立樹狀圖之前,必須先要計算距離矩陣,這裡我們以 woodmouse 資料集作為示範。
# 載入 woodmouse 資料集 data(woodmouse)
若要計算 DNA 序列資料的距離矩陣,可以使用 ape 套件所提供的 dist.dna 函數:
# 建立距離矩陣 dist.k80 <- dist.dna(woodmouse, model = "K80")
dist.dna 支援非常多種計算距離矩陣的演算法,若要使用不同的演算法可以透過 model 參數來指定。
有了距離矩陣之後,就可以呼叫 nj 函數,以近鄰結合法(neighbor-joining)產生樹狀結構:
# 以近鄰結合法建立樹狀圖 phy.k80 <- ape::nj(dist.k80)
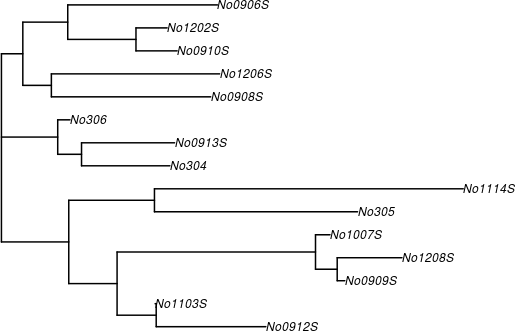
nj 所產生的樹狀結構是一個 phylo 物件,可以直接交給 plot 畫出樹狀圖:
# 繪製樹狀圖 plot(phy.k80, edge.width = 2)
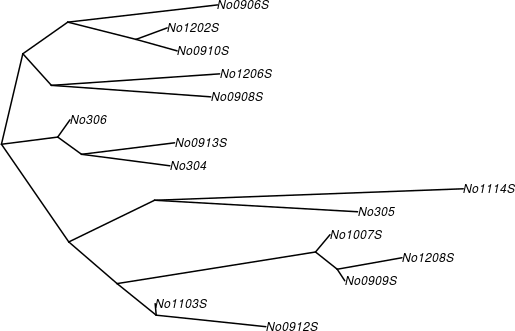
亦可畫出不同類型的樹狀圖:
# cladogram 類型的樹狀圖 plot(phy.k80, type = "cladogram", edge.width = 2)
若想要知道 plot 在繪製 phylo 的樹狀圖時可以使用的參數有哪些,可以查詢 plot.phylo 的線上說明:
?plot.phylo
若想要標記樹狀圖之中的邊或節點,可以使用 edgelabels、tiplabels 與 nodelabels 來加入文字標示。
若要將 R 中的 DNA 序列資料匯出至檔案,可以使用 ape 套件所提供的 write.dna 函數:
# 將 DNA 序列資料以 sequential 格式匯出 write.dna(woodmouse, file = "output1.txt", format = "sequential") # 將 DNA 序列資料以 interleaved 格式匯出 write.dna(woodmouse, file = "output2.txt", format = "interleaved") # 將 DNA 序列資料以 FASTA 格式匯出 write.dna(woodmouse, file = "output3.fas", format = "fasta")
write.dna 函數在處理較長的 DNA 序列資料時,速度會比較緩慢,此時建議可以改用 write.FASTA 函數:
# 將大量 DNA 序列資料以 FASTA 格式匯出 write.FASTA(woodmouse, file = "output4.fas")
參考資料:phytools.org
{kind=link}
{kind=link}